
Fixing a Baby’s Abnormal Genes in the Womb May Soon Be Possible

A couple holds up a picture of their ultrasound.
By now you have probably heard something about CRISPR, the simple and relatively inexpensive method of precisely editing the genomes of plants, animals, and humans.
The treatment of disease in fetuses, the liminal category of life between embryos and humans, poses the next frontier.
Through CRISPR and other methods of gene editing, scientists have produced crops to be more nutritious, better able to resist pests, and tolerate droughts; engineered animals ranging from fruit flies to monkeys to make them better suited for scientific study; and experimentally treated the HIV virus, Hepatitis B, and leukemia in human patients.
There are also currently FDA-approved trials to treat blindness, cancer, and sickle cell disease in humans using gene editing, and there is consensus that CRISPR's therapeutic applications will grow significantly in the coming years.
While the treatment of human disease through use of gene editing is not without its medical and ethical concerns, the avoidance of disease in embryos is far more fraught. Nonetheless, Naturereported in November that He Jiankui, a scientist in China, had edited twin embryos to disable a gene called CCR5 in hopes of avoiding transmission of HIV from their HIV-positive father.
Though there are questions about the effectiveness and necessity of this therapy, He reported that sequencing has proven his embryonic gene edits were successful and the twins were "born normal and healthy," although his claims have not been independently verified.
More recently, Denis Rebrikov, a Russian scientist, announced his plans to disable the same gene in embryos to be implanted in HIV-positive women later this year. Futuristic as it may seem, prenatal gene editing is already here.
The treatment of disease in fetuses, the liminal category of life between embryos and humans, poses the next frontier. Numerous conditions—some minor, some resulting in a lifetime of medical treatment, some incompatible with life outside of the womb—can be diagnosed through use of prenatal diagnostic testing. There is promising research suggesting doctors will soon be able to treat or mitigate at least some of them through use of fetal gene editing.
This research could soon present women carrying genetically anomalous fetuses a third option aside from termination or birthing a child who will likely face a challenging and uncertain medical future: Whether to undergo a fetal genetic intervention.
However, genetic intervention will open the door to a host of ethical considerations, particularly with respect to the relationship between pregnant women and prenatal genetic counselors. Current counselors theoretically provide objective information and answer questions rather than advise their pregnant client whether to continue with her pregnancy, despite the risks, or to have an abortion.
In practice, though, prenatal genetic counseling is most often directive, and the nature of the counseling pregnant women receive can depend on numerous factors, including their religious and cultural beliefs, their perceived ability to handle a complicated pregnancy and subsequent birth, and their financial status. Introducing the possibility of a fetal genetic intervention will exacerbate counselor reliance upon these considerations and in some cases lead to counseling that is even more directive.
Some women in the near future will face the choice of whether to abort, keep, or treat a genetically anomalous fetus.
Future counselors will have to figure out under what circumstances it is even appropriate to broach the subject. Should they only discuss therapies that are FDA-approved, or should they mention experimental treatments? What about interventions that are available in Europe or Asia, but banned in the United States? Or even in the best case of scenario of an FDA-approved treatment, should a counselor make reference to it if she knows for a fact that her client cannot possibly afford it?
Beyond the basic question of what information to share, counselors will have to confront the fact that the very notion of fixing or "editing" offspring will be repugnant to many women, and inherent in the suggestion is the stigmatization of individuals with disabilities. Prenatal genetic counselors will be on the forefront of debates surrounding which fetuses should remain as they are and which ones should be altered.
Despite these concerns, some women in the near future will face the choice of whether to abort, keep, or treat a genetically anomalous fetus in utero. Take, for example, a woman who learns during prenatal testing that her fetus has Angelman syndrome, a genetic disorder characterized by intellectual disability, speech impairment, loss of muscle control, epilepsy, and a small head. There is currently no human treatment for Angelman syndrome, which is caused by a loss of function in a single gene, UBE3A.
But scientists at the University of North Carolina have been able to treat Angelman syndrome in fetal mice by reactivating UBE3A through use of a single injection. The therapy has also proven effective in cultured human brain cells. This suggests that a woman might soon have to consider injecting her fetus's brain with a CRISPR concoction custom-designed to target UBE3A, rather than terminate her pregnancy or bring her fetus to term unaltered.
Assuming she receives the adequate information to make an informed choice, she too will face an ethical conundrum. There will be the inherent risks of injecting anything into a developing fetus's brain, including the possibility of infection, brain damage, and miscarriage. But there are also risks specific to gene editing, such as so-called off-target effects, the possibility of impacting genes other than the intended one. Such effects are highly unpredictable and can be difficult to detect. So too is it impossible to predict how altering UBE3A might lead to other genetic and epigenetic changes once the baby is born.
There are no easy answers to the many questions that will arise in this space.
A woman deciding how to act in this scenario must balance these risks against the potential benefits of the therapy, layered on top of her belief system, resources, and personal ethics. The calculus will be different for every woman, and even the same woman might change her mind from one pregnancy to the next based on the severity of the condition diagnosed and other available medical options.
Her genetic counselor, meanwhile, must be sensitive to all of these concerns in helping her make her decision, keeping up to date on the possible new treatments, and carefully choosing which information to disclose in striving to be neutral. There are no easy answers to the many questions that will arise in this space, but better to start thinking about them now, before it is too late.
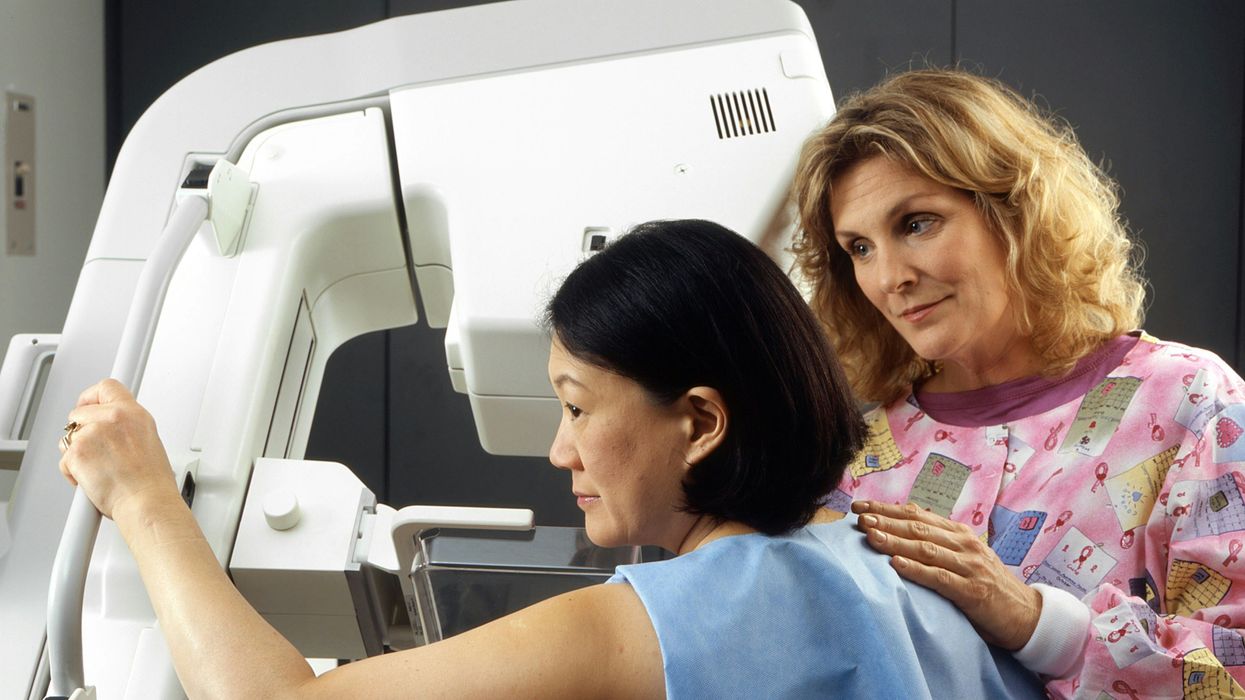
A woman receives a mammogram, which can detect the presence of tumors in a patient's breast.
When a patient is diagnosed with early-stage breast cancer, having surgery to remove the tumor is considered the standard of care. But what happens when a patient can’t have surgery?
Whether it’s due to high blood pressure, advanced age, heart issues, or other reasons, some breast cancer patients don’t qualify for a lumpectomy—one of the most common treatment options for early-stage breast cancer. A lumpectomy surgically removes the tumor while keeping the patient’s breast intact, while a mastectomy removes the entire breast and nearby lymph nodes.
Fortunately, a new technique called cryoablation is now available for breast cancer patients who either aren’t candidates for surgery or don’t feel comfortable undergoing a surgical procedure. With cryoablation, doctors use an ultrasound or CT scan to locate any tumors inside the patient’s breast. They then insert small, needle-like probes into the patient's breast which create an “ice ball” that surrounds the tumor and kills the cancer cells.
Cryoablation has been used for decades to treat cancers of the kidneys and liver—but only in the past few years have doctors been able to use the procedure to treat breast cancer patients. And while clinical trials have shown that cryoablation works for tumors smaller than 1.5 centimeters, a recent clinical trial at Memorial Sloan Kettering Cancer Center in New York has shown that it can work for larger tumors, too.
In this study, doctors performed cryoablation on patients whose tumors were, on average, 2.5 centimeters. The cryoablation procedure lasted for about 30 minutes, and patients were able to go home on the same day following treatment. Doctors then followed up with the patients after 16 months. In the follow-up, doctors found the recurrence rate for tumors after using cryoablation was only 10 percent.
For patients who don’t qualify for surgery, radiation and hormonal therapy is typically used to treat tumors. However, said Yolanda Brice, M.D., an interventional radiologist at Memorial Sloan Kettering Cancer Center, “when treated with only radiation and hormonal therapy, the tumors will eventually return.” Cryotherapy, Brice said, could be a more effective way to treat cancer for patients who can’t have surgery.
“The fact that we only saw a 10 percent recurrence rate in our study is incredibly promising,” she said.

Urinary tract infections account for more than 8 million trips to the doctor each year.
Few things are more painful than a urinary tract infection (UTI). Common in men and women, these infections account for more than 8 million trips to the doctor each year and can cause an array of uncomfortable symptoms, from a burning feeling during urination to fever, vomiting, and chills. For an unlucky few, UTIs can be chronic—meaning that, despite treatment, they just keep coming back.
But new research, presented at the European Association of Urology (EAU) Congress in Paris this week, brings some hope to people who suffer from UTIs.
Clinicians from the Royal Berkshire Hospital presented the results of a long-term, nine-year clinical trial where 89 men and women who suffered from recurrent UTIs were given an oral vaccine called MV140, designed to prevent the infections. Every day for three months, the participants were given two sprays of the vaccine (flavored to taste like pineapple) and then followed over the course of nine years. Clinicians analyzed medical records and asked the study participants about symptoms to check whether any experienced UTIs or had any adverse reactions from taking the vaccine.
The results showed that across nine years, 48 of the participants (about 54%) remained completely infection-free. On average, the study participants remained infection free for 54.7 months—four and a half years.
“While we need to be pragmatic, this vaccine is a potential breakthrough in preventing UTIs and could offer a safe and effective alternative to conventional treatments,” said Gernot Bonita, Professor of Urology at the Alta Bro Medical Centre for Urology in Switzerland, who is also the EAU Chairman of Guidelines on Urological Infections.
The news comes as a relief not only for people who suffer chronic UTIs, but also to doctors who have seen an uptick in antibiotic-resistant UTIs in the past several years. Because UTIs usually require antibiotics, patients run the risk of developing a resistance to the antibiotics, making infections more difficult to treat. A preventative vaccine could mean less infections, less antibiotics, and less drug resistance overall.
“Many of our participants told us that having the vaccine restored their quality of life,” said Dr. Bob Yang, Consultant Urologist at the Royal Berkshire NHS Foundation Trust, who helped lead the research. “While we’re yet to look at the effect of this vaccine in different patient groups, this follow-up data suggests it could be a game-changer for UTI prevention if it’s offered widely, reducing the need for antibiotic treatments.”