
Is Alzheimer's Research On the Wrong Track?

Scientist in laboratory
"The graveyard of hope." That's what experts call the quest for effective Alzheimer's treatments, a two-decade effort that has been marked by one costly and high-profile failure after another. Nearly all of the drugs tested target one of the key hallmarks of Alzheimer's disease: amyloid plaques, the barnacle-like proteins long considered the culprits behind the memory-robbing ravages of the disease. Yet all the anti-amyloid drugs have flopped miserably, prompting some scientists to believe we've fingered the wrong villain.
"We're flogging a dead horse," says Peter Davies, PhD, an Alzheimer's researcher at the Feinstein Institute for Medical Research in New York. "The fact that no one's gotten better suggests that you have the wrong mechanism."
If the naysayers are right, how could a scientific juggernaut of this magnitude—involving hundreds of scientists in academia and industry at a cost of tens of billions of dollars--be so far off the mark? There are no easy answers, but some experts believe this calls into question how research is conducted and blame part of the failure on the insular culture of the scientific aristocracy at leading academic institutions.
"The field began to be dominated by narrow views."
"The field began to be dominated by narrow views," says George Perry, PhD, an Alzheimer's researcher and dean of the College of Sciences at the University of Texas in San Antonio. "The people pushing this were incredibly articulate, powerful and smart. They'd go to scientific meetings and all hang around with each other and they'd self-reinforce."
In fairness, there was solid science driving this. Post-mortem analyses of Alzheimer's patients found their brains were riddled with amyloid plaques. People with a strong family history of Alzheimer's had genetic mutations in the genes that encode for the production of amyloids. And in animal studies, scientists found that if amyloids were inserted into the brains of transgenic mice, they exhibited signs of memory loss. Remove the amyloids and they suddenly got better. This body of research helped launch the Amyloid Cascade Hypothesis of the disease in 1992—which has driven research ever since.
Scientists believed that the increase in the production of these renegade proteins, which form sticky plaques and collect outside of the nerve cells in the brain, triggers a series of events that interfere with the signaling system between synapses. This seems to prevent cells from relaying messages or talking to each other, causing memory loss, confusion and increasing difficulties doing the normal tasks of life. The path forward seemed clear: stop amyloid production and prevent disease progression. "We were going after the obvious abnormality," says Dr. David Knopman, a neurologist and Alzheimer's researcher at the Mayo Clinic in Rochester, Minnesota.
"Why wouldn't you do that?" Why ideed.
In hindsight, though, there was no real smoking gun—no one ever showed precisely how the production of amyloids instigates the destruction of vital brain circuits.
"Amyloids are clearly important," says Perry, "but they have not proven to be necessary and sufficient for the development of this disease."
Ironically, there have been hints all along that amyloids may not be toxic bad boys.
A handful of studies revealed that amyloid proteins are produced in healthy brains to protect synapses. Research on animal models that mimic diseases suggest that certain forms of amyloids can ease damage from strokes, traumatic brain injuries and even heart attacks. In a 2013 study, to cite just one example, a Stanford University team injected synthetic amyloids into paralyzed mice with an inflammatory disorder similar to multiple sclerosis. Instead of worsening their symptoms—which is what the researchers expected to happen--the mice could suddenly walk again. Remove the amyloids, and they became paralyzed once more.
Still other studies suggest amyloids may actually function as molecular guardians dispatched to silence inflammation and mop up errant cells after an injury as part of the body's waste management system. "The presence of amyloids is a protective response to something going wrong, a threat," says Dr. Dale Bredesen, a UCLA neurologist. "But the problem arises when the threats are chronic, multiple, unrelenting and intense. The defenses the brain mounts are also intense and these protective mechanisms cross the line into causing harm, and killing the very synapses and brain cells the amyloid was called up to protect."
So how did research get derailed?
In a way, we're victims of our own success, critics say.
Early medical triumphs in the heady post-World War II era, like the polio vaccine that eradicated the crippling childhood killer, or antibiotics, reinforced the magic bullet idea of curing disease--find a target and then hit it relentlessly. That's why when scientists made the link between amyloids and disease progression, Big Pharma jumped on the bandwagon in hopes of inventing a trillion-dollar drug. This approach is fine when you have an acute illness, like an infectious disease that's caused by one agent, but not for something as complicated as Alzheimer's.
The other piece of the problem is the dwindling federal dollars for basic research. Maverick scientists find it difficult to secure funding, which means that other possible targets or approaches remained relatively unexplored—and drug companies are understandably reluctant to sponsor fishing expeditions with little guarantee of a payoff. "Very influential people were driving this hypothesis," says Davies, and with careers on the line, "there was not enough objectivity or skepticism about that hypothesis."
Still, no one is disputing the importance of anti-amyloid drugs—and ongoing clinical trials, like the DIAN and A4 studies, are intervening earlier in patients who are at a high risk of developing Alzheimer's, but before they're symptomatic. "The only way to know if this is really a dead end is if you take it as far as it can go," says Knopman. "I believe the A4 study is the proper way to test the amyloid hypothesis."
But according to some experts, the latest thinking is that Alzheimer's is triggered by a range of factors, including genetics, poor diet, stress and lack of exercise.
"Alzheimer's is like other chronic age-related diseases and is multi-factorial," says Perry. "Modulating amyloids may have value but other avenues need to be explored."
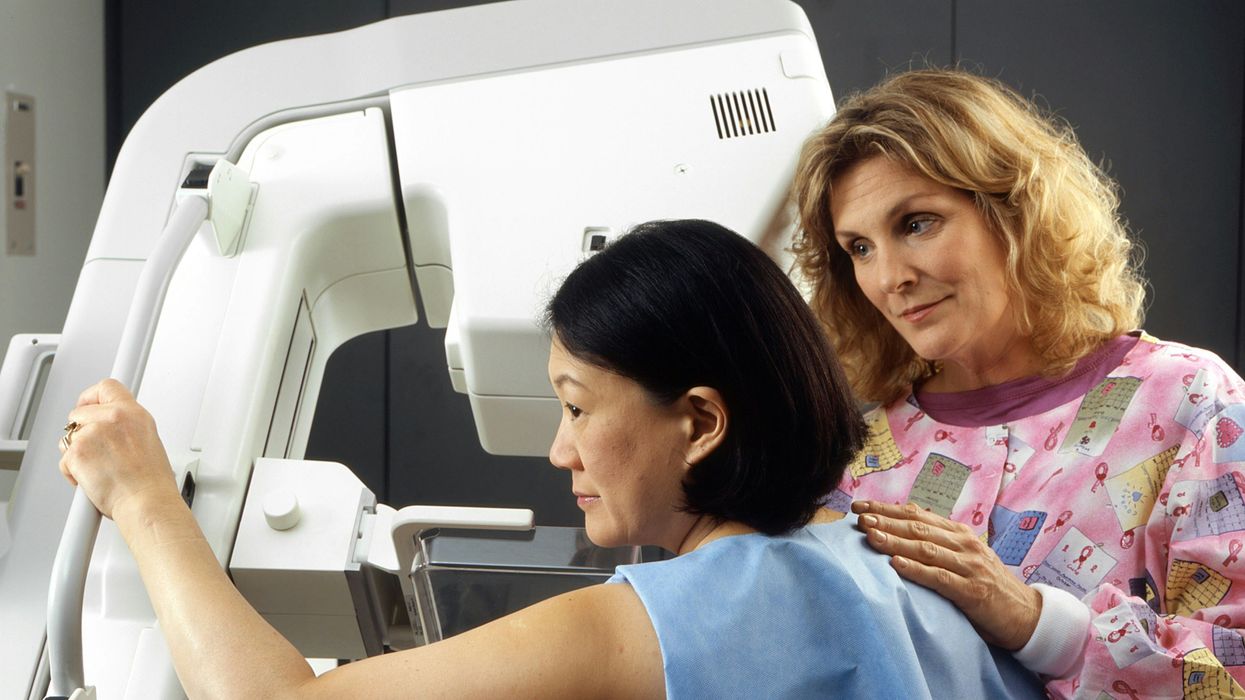
A woman receives a mammogram, which can detect the presence of tumors in a patient's breast.
When a patient is diagnosed with early-stage breast cancer, having surgery to remove the tumor is considered the standard of care. But what happens when a patient can’t have surgery?
Whether it’s due to high blood pressure, advanced age, heart issues, or other reasons, some breast cancer patients don’t qualify for a lumpectomy—one of the most common treatment options for early-stage breast cancer. A lumpectomy surgically removes the tumor while keeping the patient’s breast intact, while a mastectomy removes the entire breast and nearby lymph nodes.
Fortunately, a new technique called cryoablation is now available for breast cancer patients who either aren’t candidates for surgery or don’t feel comfortable undergoing a surgical procedure. With cryoablation, doctors use an ultrasound or CT scan to locate any tumors inside the patient’s breast. They then insert small, needle-like probes into the patient's breast which create an “ice ball” that surrounds the tumor and kills the cancer cells.
Cryoablation has been used for decades to treat cancers of the kidneys and liver—but only in the past few years have doctors been able to use the procedure to treat breast cancer patients. And while clinical trials have shown that cryoablation works for tumors smaller than 1.5 centimeters, a recent clinical trial at Memorial Sloan Kettering Cancer Center in New York has shown that it can work for larger tumors, too.
In this study, doctors performed cryoablation on patients whose tumors were, on average, 2.5 centimeters. The cryoablation procedure lasted for about 30 minutes, and patients were able to go home on the same day following treatment. Doctors then followed up with the patients after 16 months. In the follow-up, doctors found the recurrence rate for tumors after using cryoablation was only 10 percent.
For patients who don’t qualify for surgery, radiation and hormonal therapy is typically used to treat tumors. However, said Yolanda Brice, M.D., an interventional radiologist at Memorial Sloan Kettering Cancer Center, “when treated with only radiation and hormonal therapy, the tumors will eventually return.” Cryotherapy, Brice said, could be a more effective way to treat cancer for patients who can’t have surgery.
“The fact that we only saw a 10 percent recurrence rate in our study is incredibly promising,” she said.

Urinary tract infections account for more than 8 million trips to the doctor each year.
Few things are more painful than a urinary tract infection (UTI). Common in men and women, these infections account for more than 8 million trips to the doctor each year and can cause an array of uncomfortable symptoms, from a burning feeling during urination to fever, vomiting, and chills. For an unlucky few, UTIs can be chronic—meaning that, despite treatment, they just keep coming back.
But new research, presented at the European Association of Urology (EAU) Congress in Paris this week, brings some hope to people who suffer from UTIs.
Clinicians from the Royal Berkshire Hospital presented the results of a long-term, nine-year clinical trial where 89 men and women who suffered from recurrent UTIs were given an oral vaccine called MV140, designed to prevent the infections. Every day for three months, the participants were given two sprays of the vaccine (flavored to taste like pineapple) and then followed over the course of nine years. Clinicians analyzed medical records and asked the study participants about symptoms to check whether any experienced UTIs or had any adverse reactions from taking the vaccine.
The results showed that across nine years, 48 of the participants (about 54%) remained completely infection-free. On average, the study participants remained infection free for 54.7 months—four and a half years.
“While we need to be pragmatic, this vaccine is a potential breakthrough in preventing UTIs and could offer a safe and effective alternative to conventional treatments,” said Gernot Bonita, Professor of Urology at the Alta Bro Medical Centre for Urology in Switzerland, who is also the EAU Chairman of Guidelines on Urological Infections.
The news comes as a relief not only for people who suffer chronic UTIs, but also to doctors who have seen an uptick in antibiotic-resistant UTIs in the past several years. Because UTIs usually require antibiotics, patients run the risk of developing a resistance to the antibiotics, making infections more difficult to treat. A preventative vaccine could mean less infections, less antibiotics, and less drug resistance overall.
“Many of our participants told us that having the vaccine restored their quality of life,” said Dr. Bob Yang, Consultant Urologist at the Royal Berkshire NHS Foundation Trust, who helped lead the research. “While we’re yet to look at the effect of this vaccine in different patient groups, this follow-up data suggests it could be a game-changer for UTI prevention if it’s offered widely, reducing the need for antibiotic treatments.”